|
講義内容
はじめに吉井先生に分子シミュレーションの基礎ついてお話しいただいた後、
部屋を分けて篠田先生と山内先生にそれぞれ講義して頂きます。
|
| 講師 |
吉井範行 先生(名古屋大学)
|
| タイトル |
分子シミュレーションの基礎
|
| 講演概要 |
本講義では分子シミュレーション、とりわけ古典系の分子動力学(MD)計算を行い研究を進めるうえで必要となる一通りの方法論について説明します。
MD計算は、分子の運動を扱うという意味では古典力学・解析力学を基礎としています。また温度や圧力の制御あるいは計算結果の解析を行うためには熱力学・統計力学が不可欠であり、静電相互作用のような長距離力を取り扱うためには電磁気学を知らなければなりません。さらに運動方程式を数値的に取り扱うためには数値計算法についても精通している必要があります。このようにさまざまな学問分野の上に成り立つMD計算について、学術的基礎から実際に計算で用いる表式まで導きたいと思います。
講義内容は以下の通りです。
1.MD計算のあらまし
2.運動方程式の一般的な表し方 ─並進・回転運動を記述するための基礎
3.運動方程式の数値計算法 ─数値的に解くための基礎
4.長距離力の取り扱い方 ─エワルドの方法と高速多重極展開法(FMM)
5.アンサンブルの発生 ─温度と圧力を制御する方法
6.拘束条件つきの運動方程式の解き方 ─拘束の動力学の数値計算法
7.自由エネルギー計算法の原理 ─化学現象を理解するための最も重要な熱力学量
8.MD計算から求められる物理量 ─静的・動的な性質を求めるための基礎
|
| 講師 |
篠田渉 先生(産業技術総合研究所)
| 
|
| タイトル |
自己組織化膜の分子動力学計算
|
| 講演概要 |
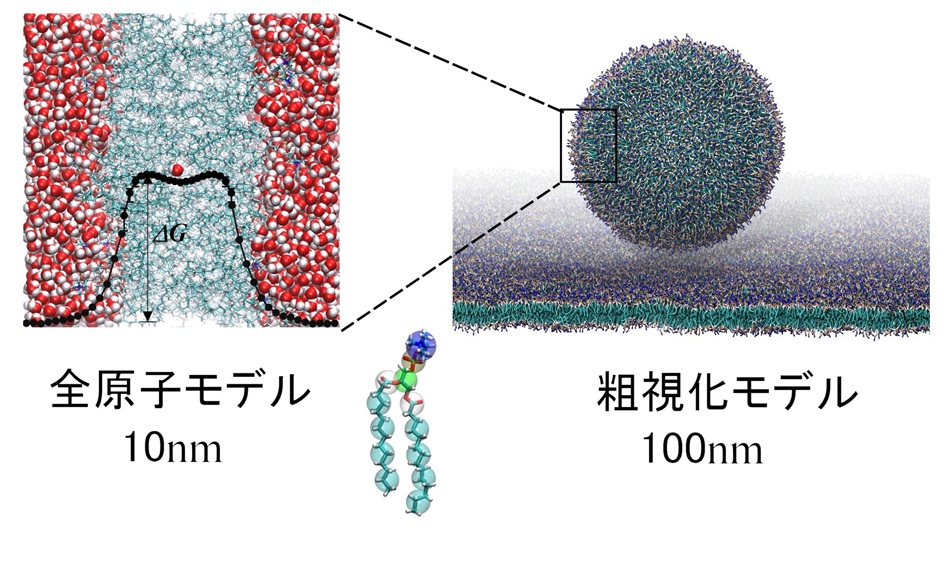
脂質や界面活性剤のような両親媒性分子は溶媒中で様々な形態の自己組織化構造を形成する。この中で特に脂質の作る二重層膜構造は生体膜モデルとして長く研究されてきた。本講義では自己組織化膜の分子動力学シミュレーション(MD)の現状について、分子モデリングや方法論の課題を交えて2部構成で紹介する。
第1部では、全原子モデルに基づいた脂質二重層膜のMDを紹介する。境界条件や力場などMDにおける問題を整理した後、分子透過性の評価手法(自由エネルギー解析)などについて解説を行う。
第2部では、粗視化モデリング手法を概説し、分子集合構造形態の定量解析を可能とする粗視化法について述べる。様々な適用研究例を紹介するとともに、マルチスケールの分子シミュレーションの利点について解説する。さらに時間が許せば連続体膜弾性理論との接合における課題について述べたい。
|
| 講師 |
山内淳 先生(慶應義塾大学)
| 
|
| タイトル |
第一原理計算の基礎:密度汎関数法を中心として
|
| 講演概要 |
夏の学校のタイトルにもあります「分子シミュレーション」を行うにあたり、分子あるいは原子のようなミクロな対象に働く力は電子が介在することによって生じます。電子の振る舞いをきちんと理解するためには量子力学が必要になってくるわけですが、残念ながら量子力学を真っ正面から解くことは非常に難しく、事実上不可能です。というわけで、実用上は、実験(あるいは高精度な量子力学的結果)を再現する様にパラメタを調整したモデル(ポテンシャル)を使用して研究することが多くなります。しかしながら、このようなモデルはパラメタを決定した原子配置に大きく依存して汎用性が限られてしまいます。
一方で、実験のパラメタなどを用いずに、量子力学のみを基礎として電子状態を効率的に求めようとする方法論が第一原理計算と呼ばれる手法です。いくつか種類があり、その内の一つがこの講義のテーマである密度汎関数法です。一言で言うと、問題設定を変えることによって、主役を波動関数から、電子密度に置き換えることによって計算の効率化を実現しています。第一原理計算分野の他の方法と比べると計算効率が非常に高いので、広く使われています。
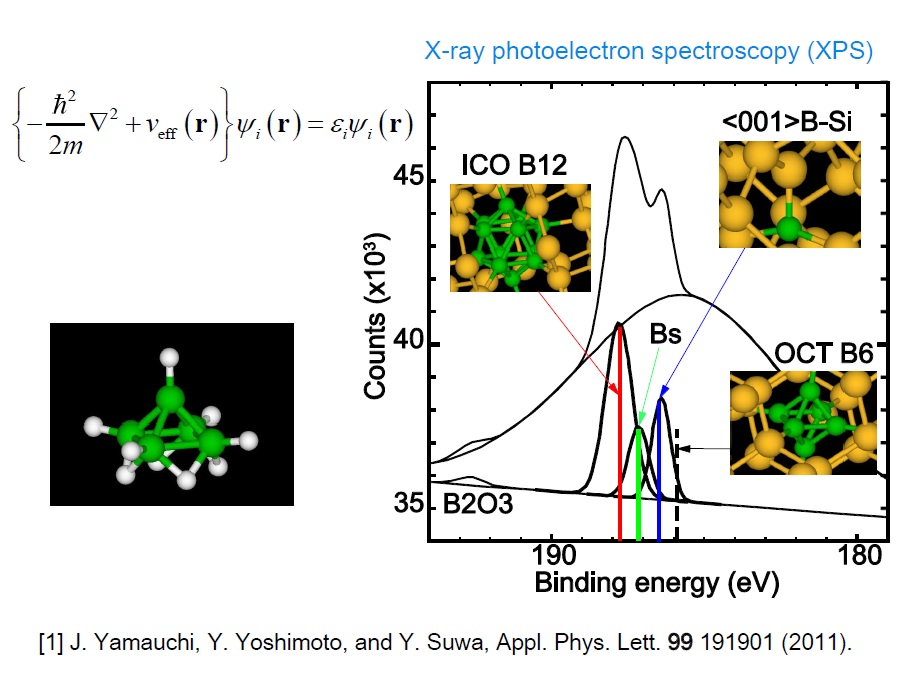
講義では、量子力学を厳密に解くことが何故難しいのかというところから始めて、密度汎関数法の基礎について説明していきます。また最近は計算機の進歩も著しくパッケージソフト等も出回っていますから、それらを使うときの基礎となる概念についても説明したいと考えています。最後に、応用例として、モデルポテンシャルとして取り扱うのに難しい元素であるホウ素(B)のシリコン結晶中の多彩な振る舞いについてお話しする予定です。
|
|
|
|